
Electron Microscopy Tutorial
Overview
Electron microscopes use electrons to illuminate a sample. In Transmission Electron Microscopy (TEM), electrons pass through the sample and illuminate film or a digital camera. Electron dense material in the sample casts shadows on the camera face and thereby produces a two-dimensional projection of material in the section.
Wavelength and Resolution
Resolution in microscopy is limited to about ½ of the wavelength of the illumination source used to image the sample (see Super-resolution Tutorial). Because our eyes can only detect photons with wavelengths greater than ~400 nm, the best resolution that can be achieved by light microscopes is about ~200 nm. One way to beat the diffraction limit of light is to use an illumination source with a shorter wavelength than photons – electrons (see Box 1).
Box 1: The de Broglie relationship
Louis de Broglie showed that every particle or matter propagates like a wave. The wavelength of a particle or a matter can be calculated as follows.
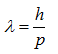
where λ is the wavelength of a particle, h is Planck’s constant (6.626 x 10-34 J seconds), and p is the momentum of a particle. Since the momentum is the product of the mass and the velocity of a particle,
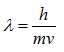
Because the velocity of the electrons is determined by the accelerating voltage, or electron potential where
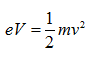
The velocity of electrons can be calculated by
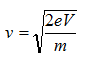
Therefore, the wavelength of propagating electrons at a given accelerating voltage can be determined by
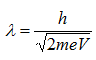
Since the mass of an electron is 9.1 x 10-31 kg and e = 1.6 x 10-19,
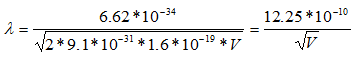
Thus, the wavelength of electrons is calculated to be 3.88 pm when the microscope is operated at 100 keV, 2.74 pm at 200 keV, and 2.24 pm at 300 keV.
However, because the velocities of electrons in an electron microscope reach about 70% the speed of light with an accelerating voltage of 200 keV, there are relativistic effects on these electrons. These effects include significant length contraction, time dilation, and an increase in mass. By accounting for these changes,
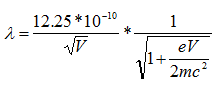
where c is the speed of light, which is ~3 x 108 m/s. Therefore, the wavelength at 100 keV, 200 keV, and 300 keV in electron microscopes is 3.70 pm, 2.51 pm and 1.96 pm, respectively.
The wavelength of electrons is much smaller than that of photons (2.5 pm at 200 keV). Thus the resolution of an electron microscope is theoretically unlimited for imaging cellular structure or proteins. Practically, the resolution is limited to ~0.1 nm due to the objective lens system in electron microscopes. Thus, electron microscopy can resolve subcellular structures that could not be visualized using standard fluorescences microscopy, such as the microvilli of intestinal cells or the internal structure of a bacterium (Figure 1).
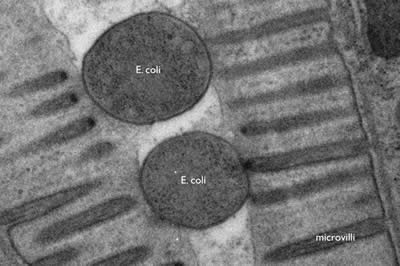
Figure 1: E. coli bacteria being digested in the intestine of a nematode, C. elegans. The microvilli of the intestinal cells project into the lumen of the gut; the bacteria are lodged in the lumen. The cross-section of E. coli is about 400 nm. (photo Shigeki Watanabe and Erik Jorgensen)
Electron microscopes can even resolve molecular structure in a cell, for example the plasma membrane is only about 5nm in diameter. The membrane is composed of a lipid bilayer, each layer is a single molecule thick. These fatty acids are about 20 carbons long with a hydrophilic head group. These individual lipid layers can be distinguished in an electron micrograph (Figure 2).
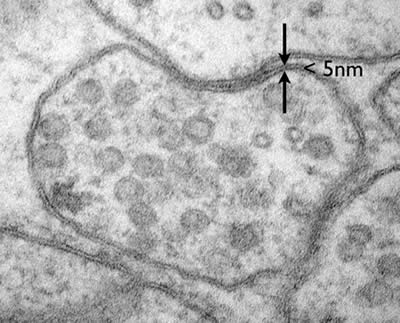
Figure 2: A neuromuscular junction in C. elegans. The lipid bilayer of the plasma membrane is less than 5nm but the individual leaflets of the bilayer can be resolved. (photo Shigeki Watanabe and Erik Jorgensen)
Fixation
For transmission electron microscopy, a beam of electrons is passed through a sample. Because electron microscopes are operated in a vacuum, specimens need to be dehydrated. To avoid alterations in tissues caused by dehydration (like raisins from grapes), the tissue must be cross-linked or ‘fixed’ to preserve structure. Fixatives typically used in electron microscopy include glutaraldehyde or osmium tetroxide. Conventionally fixation is performed on ice since tissue preservation is better in immobilized and inactive samples.
High-Pressure Freezing
Conventional chemical fixation often leads to artifacts such as shrinkage, membrane distortion and aggregation of proteins. Rapid freezing is a widely used method for stopping cellular metabolism and activity instantaneously and can avoid some of the artifacts observed using ice-cold fixatives. Freezing of specimens, however, introduces new artifacts because the water in cells becomes crystallized during the process. Ice expands and breaks membranes. To avoid the formation of ice crystals, samples can be frozen under high-pressure (~2000 bar). Such samples exhibit excellent preservation (Figure 3).
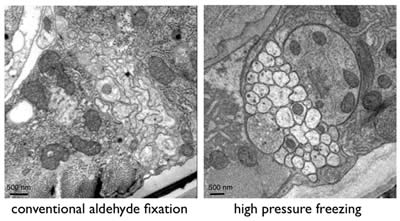
Figure 3: Ventral nerve cord of C. elegans showing the differences between conventional chemical fixation and high-pressure freezing.
Normally when liquid water is cooled to freezing temperature, water molecules begins to form ice on a seed crystal or some other nucleating structure (Figure 4B). Since water expands when it crystallizes, it can break cellular membranes (Figure 4E). In the absence of a nucleation site, super-cooled water can stay in its liquid phase (Figure 4C). Eventually, the liquid will reach its homogeneous nucleation state and crystallize in the absence of a nucleus. The temperature at which homogeneous nucleation takes place for water is around -40ºC. If the freezing rate of 10,000ºC/s can be achieved, super-cooled water can be vitrified without crystallization, that is, the water will freeze in an unordered state (Figure 4C). Liquid nitrogen can generate a cooling rate of -16,000ºC/s. However due to the poor heat conductance of water, the freezing rate of tissue 10µm deep is very slow, and therefore thick samples (>10 µm) cannot be frozen without ice crystals. However, at 2100 bar (1 bar = atmospheric pressure at sea level), water can be super-cooled to -90ºC. Under these conditions, a freezing rate of 200ºC/s is sufficient to vitrify water. Thus, despite the poor heat conductance of water, biological tissues as thick as 500 µm can be frozen without ice crystals (Figure 4F). Intact animals such as C. elegans and Drosophila larva as well as tissue slices can be frozen and preserved for electron microscopy.
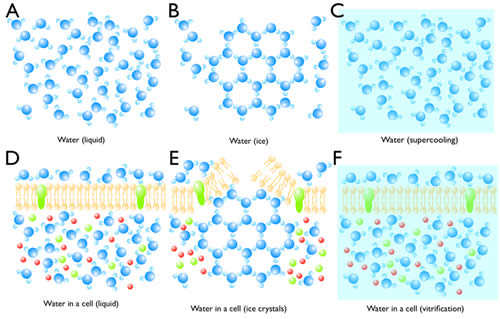
Figure 4: Crystallization and vitrification of water. (A) Water molecules are highly mobile in liquid. (B) When the temperature reaches 0ºC, water starts to crystallize if there is a seed. (C) Water can be super-cooled to -40ºC at atmospheric pressure. (D) Water in a cell is also highly mobile despite the presence of molecules in the cytosol. (E) Ice crystals can break the cell membrane and also displace molecules and cellular structures. (F) By high-pressure freezing, water vitrifies, thereby preserving the native state of a cell.
Sectioning
After fixation, the sample is embedded in plastic and thin sections cut on a diamond knife using a microtome (Figure 5). Since electrons must pass through a specimen to form an image, tissues must be sliced into 30-80 nm sections for TEM imaging. A microtome advances the specimen against a stationary diamond knife with nanometer precision by using the thermal expansion of metals. The quality of sections depends on three factors: the environment, embedding plastic, and knife. A microtome must be situated in a room where there is no vibration or thermal fluctuations. The thickness of sections can be easily altered by vibration or temperature change. The plastic in which the specimens are embedded also can affect section quality. There are several types of plastics commercially available and they are either epoxy-based (Araldite, Durcupan, Epon, and Spurr) or acrylic-based (LR white, methacrylates, and Lowicryl) plastics. Epoxy resins are polymerized using a catalyst and high temperature. The advantage of epoxy-based resins is that they cross-link with tissues and form ribbons nicely. On the other hand, they are very hydrophobic, and thus are not suitable for immunocytochemistry. Acrylic resins are polymerized using either UV-light, a catalyst or high temperature. The properties of acrylic resins are the opposite of expoxy resins: because the tissue does not crosslink with the resin, the sections often suffer ‘drop-outs’, the hygroscopic nature can lead to expansion and poor ribbon formation. On the other hand the hydrophilic nature makes these reasons suitable for immunocytochemistry. Thus, depending on the application, a different type of resin can be employed. Finally, a sharp knife is imperative for a good quality of sectioning. Three types of knives are commercially available: glass (least expensive), sapphire, and diamond (most expensive). All of these knives can be used for ultrathin sectioning. However, the durability of an edge of a diamond knife is the highest, and thereby many sections can be sectioned with the same quality. Knife choice will depend on one’s budget and usage needs.
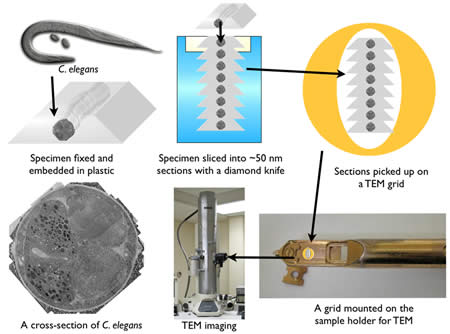
Figure 5 : A sequence for the specimen preparation for TEM.
Imaging
How can an electron be used to image structure? Electrons are negatively charged particles, and are repelled by electrons surrounding the nucleus of atoms. This interaction causes propagating electrons to scatter. Thus, contrast can be generated by electron-dense materials in the path of the electron beam. There are two factors affecting scattering in transmission electron microscopy: sample thickness and the atomic number of the contrasting agent (Figure 6).
The penetration depth of electrons depends on the section thickness of the specimen. By applying a high-voltage driving force, electrons will penetrate through tissue sections with very little scattering. Typically, ~100-150 nm is the thickest sample that can be observed using a 100 keV accelerating voltage.
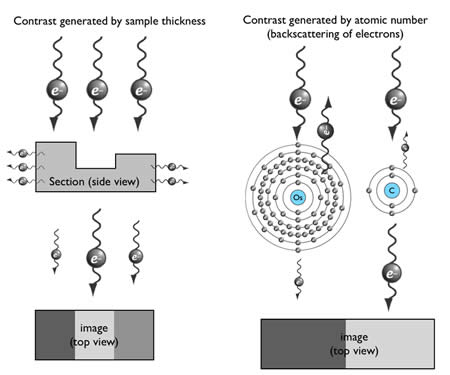
Figure 6: Generation of contrast in TEM imaging
The generation of contrast depends on the atomic number of atoms. Atoms with high atomic number possess more electrons around their nucleus, and thus more incident electrons will be scattered (Figure 6). Unfortunately, biological specimens do not generate much contrast because they are typically composed of carbon, nitrogen, and oxygen – atoms with similar atomic number. Thus tissues are stained with heavy metals, such as osmium, uranium or lead. For example, osmium preferentially binds to the unsaturated bond in lipid bilayers and crosslinks it with a neighboring lipid, and thus osmium is used to enhance contrast of membranes.
Imaging proteins
A problem with electron microscopy is that it is not very well-suited for the identification of proteins in sections. Immuno-electron microscopy is difficult (Figure 7), and morphology is compromised. New methods are being developed which combine super-resolution fluorescence microscopy and electron microscopy (Figure 8).
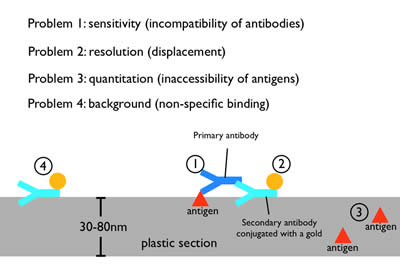
Figure 7: Immuno-electron microscopy. Moderate fixations can preserve the antigenicity of proteins during fixation and embedding. A primary antibody against the protein of interest is applied to the plastic section, followed by secondary antibody that has been conjugated with a gold particle. The gold particle is electron dense and appears as a distinct dot on the micrograph. However, the need to preserve tissue morphology restricts the use of mild fixatives for any EM preparation. Thus, antibodies that work well for fluorescence microscopy or on Western blots often do not work on plastic sections, probably due to the harsher fixation and plastic matrix (1). Moreover, each antibody is ~19 nm in length, and thus the gold particle will be displaced from the protein by up to two antibody lengths (2). Since a plastic section is 30-80 nm in thickness, some of the proteins will be deep in the plastic. The primary antibody cannot penetrate deeply into plastic to label these proteins (3). Finally, secondary antibodies can stick nonspecifically to the section and lead to background staining (4).
Figure 8: Correlative fluorescence and electron microscopy. In this image a mitochondrial protein (Tom20) was tagged with citrine and fluoresecence was imaged using STED microscopy (STimulated Emission Depletion). The section was then imaged on a scanning electron microscope to visualize organelles. The outer membrane of mitochondria can be seen in the muscle cell of a C. elegans adult. (Photo: Punge and Hell; Watanabe and Jorgensen).